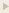In a new study, Sidman et al. report the inhibitory effect of a peptide drug called Vasotide on blood vessel overgrowth in the retinas of three animal models of human blinding retinal diseases (two rodent and one nonhuman primate). Delivery of Vasotide in eye drops prevented a blood vessel growth-promoting molecule, VEGF, from binding to two different receptors, VEGF receptor-1 and neuropilin-1, expressed by the retinal endothelial cells that line the inner surface of blood vessels.
IB4-stained branched vessels and tufts in horizontal retinal scans in normal wildtype mice and mice with oxygen-induced retinopathy treated with a control peptide, D(CAPAC), or the drug Vasotide.
Figure Credit: Sidman R.L. et al., Science Translational Medicine (2015)
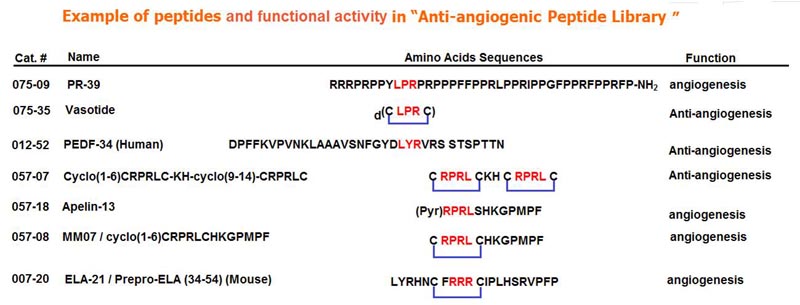
Blood vessel growth from preexisting vessels (angiogenesis) underlies many severe diseases including major blinding retinal diseases such as retinopathy of prematurity (ROP) and aged macular degeneration (AMD). This observation has driven development of antibody inhibitors that block a central factor in AMD, vascular endothelial growth factor (VEGF), from binding to its receptors VEGFR-1 and mainly VEGFR-2. However, some patients are insensitive to current anti-VEGF drugs or develop resistance, and the required repeated intravitreal injection of these large molecules is costly and clinically problematic. We have evaluated a small cyclic retro-inverted peptidomimetic, D(Cys-Leu-Pro-Arg-Cys) [D(CLPRC)], and hereafter named Vasotide, that inhibits retinal angiogenesis by binding selectively to the VEGF receptors VEGFR-1 and neuropilin-1 (NRP-1). Delivery of Vasotide via either eye drops or intraperitoneal injection in a laser-induced monkey model of human wet AMD, a mouse genetic knockout model of the AMD subtype called retinal angiomatous proliferation (RAP), and a mouse oxygen-induced model of ROP decreased retinal angiogenesis in all three animal models. This prototype drug candidate is a promising new dual receptor inhibitor of the VEGF ligand with potential for translation into safer, less-invasive applications to combat pathological angiogenesis in retinal disorders.
Richard L. Sidman, Jianxue Li, Matthew Lawrence et al., Science Translational Medicine 2015 Oct;7(309): pp. 309ra165 DOI: 10.1126 /scitranslmed.aac4882.
[Pyr(1)]apelin-13 is an endogenous vasodilator and inotrope but is downregulated in pulmonary hypertension and heart failure, making the apelin receptor an attractive therapeutic target. Agonists acting at the same G-protein-coupled receptor can be engineered to stabilize different conformational states and function as biased ligands, selectively stimulating either G-protein or β-arrestin pathways. We used molecular dynamics simulations of apelin/receptor interactions to design cyclic analogues and identified MM07 as a biased agonist. In β-arrestin and internalization assays (G-protein-independent), MM07 was 2 orders of magnitude less potent than [Pyr(1)]apelin-13. In a G-protein-dependent saphenous vein contraction assay, both peptides had comparable potency (pD2:[Pyr(1)]apelin-13 9.93±0.24; MM07 9.54±0.42) and maximum responses with a resulting bias for MM07 of ≈350- to 1300-fold for the G-protein pathway. In rats, systemic infusions of MM07 (10-100nmol) caused a dose-dependent increase in cardiac output that was significantly greater than the response to [Pyr(1)]apelin-13. Similarly, in human volunteers, MM07 produced a significant dose-dependent increase in forearm blood flow with a maximum dilatation double that is seen with [Pyr(1)]apelin-13. Additionally, repeated doses of MM07 produced reproducible increases in forearm blood flow. These responses are consistent with a more efficacious action of the biased agonist. In human hand vein, both peptides reversed an established norepinephrine constrictor response and significantly increased venous flow. Our results suggest that MM07 acting as a biased agonist at the apelin receptor can preferentially stimulate the G-protein pathway, which could translate to improved efficacy in the clinic by selectively stimulating vasodilatation and inotropic actions but avoiding activating detrimental β-arrestin-dependent pathways.
Brame AL, Maguire JJ, Yang P et al., Hypertension. 2015 Apr;65(4):834-40. doi: 10.1161/HYPERTENSIONAHA.114.05099. Epub 2015 Feb 23.
BACKGROUND: PR39 is a proline- and arginine-rich peptide implicated in wound healing and myocardial ischemia protection. To determine the potential mechanisms of PR39 in ischemia, we examined the role of PR39 in hypoxia-induced apoptosis in vascular endothelial cells.
METHODS AND RESULTS: Hypoxia results in an increase of apoptosis in bovine aortic endothelial cells (BAECs), as determined by terminal deoxynucleotidyl transferase-mediated dUTP biotin nick-end labeling (TUNEL) analysis and caspase-3 activity. Hypoxia induced 66.2+/-2.7% TUNEL-positive cells, whereas in the presence of synthesized PR39 peptide, TUNEL-positive cells were reduced to 29.6+/-1.9% (P<0.05). After 24 hours of hypoxia, the addition of PR39 reduced caspase-3 activity to 3.17+/-0.47 pMol/min from 10.52+/-0.55 pMol/min in hypoxic BAECs. Moreover, PR39 increased inhibitor of apoptosis protein-2 (IAP-2) gene and protein expression by 3-fold in a time- and dose-dependent manner. The induction of IAP-2 by PR39 conferred an increase in IAP-2 gene transcription and IAP-2 mRNA stability. Furthermore, inhibiting IAP-2 with second mitochondria-derived activator of caspase (Smac) and with small interfering RNA targeting IAP-2 abrogated the ability of PR39 to reduce caspase-3 activity.
CONCLUSIONS: We provide the first direct evidence for PR39 as an antiapoptotic factor in endothelial cells during hypoxia. These data suggest thatPR39 inhibits hypoxia-induced apoptosis and decreases caspase-3 activity in endothelial cells through an increase of IAP-2 expression.
Wu J, Parungo C, Wu G et al., Circulation. 2004 Apr 6;109(13):1660-7. Epub 2004 Mar 15.
Although tissue injury and inflammation are considered essential for the induction of angiogenesis, the molecular controls of this cascade are mostly unknown. Here we show that a macrophage-derived peptide, PR39, inhibited the ubiquitin-proteasome-dependent degradation of hypoxia-inducible factor-1alpha protein, resulting in accelerated formation of vascular structures in vitro and increased myocardial vasculature in mice. For the latter, coronary flow studies demonstrated that PR39-induced angiogenesis resulted in the production of functional blood vessels. These findings show that PR39 and related compounds can be used as potent inductors of angiogenesis, and that selective inhibition of hypoxia-inducible factor-1alpha degradation may underlie the mechanism of inflammation-induced angiogenesis.
Li J, et al. Nat Med. 2000 Jan;6(1):49-55
The rat zitter (zi) mutation induces hypomyelination and vacuolation in the central nervous system (CNS), which result in early-onset tremor and progressive flaccid paresis. By positional cloning, we found a marked decrease in Attractin (Atrn) mRNA in the brain of the zi/zi rat and identified zi as an 8-bp deletion at a splice donor site of Atrn. Atrn has been known to play multiple roles in regulating physiological processes that are involved in monocyte-T cell interaction, agouti-related hair pigmentation, and control of energy homeostasis. Rat Atrn gene encoded two isoforms, a secreted and a membrane form, as a result of alternative splicing. The zi mutation at the Atrn locus darkened coat color when introduced into agouti rats, as also described in mahogany (mg) mice, carrying the homozygous mutation at the Atrn locus. Transgenic rescue experiments showed that the membrane-type Atrn complemented both neurological alteration and abnormal pigmentation in zi/zi rats, but that the secreted-type Atrn complemented neither mutant phenotype. Furthermore, we discovered that mg mice exhibited hypomyelination and vacuolation in the CNS associated with body tremor. We conclude from these results that the membrane Atrn has a critical role in normal myelination in the CNS and would provide insights into the physiology of myelination as well as the etiology of myelin diseases.
Kuramoto T, Kitada K, Inui T et al., Proc Natl Acad Sci U S A. 2001 Jan 16;98(2):559-64.
The apelin receptor (APJ) is a class A G-protein-coupled-receptor (GPCR) and is a putative target for the treatment of cardiovascular and metabolic diseases. Apelin-13 (NH2-QRPRLSHKGPMPF-COOH) is a vasoactive peptide and one of the most potent endogenous inotropic agents identified to date. We report the design and discovery of a novel APJ antagonist. By using a bivalent ligand approach, we have designed compounds with two 'affinity' motifs and a short series of linker groups with different conformational and non-bonded interaction properties. One of these, cyclo(1-6)CRPRLC-KH-cyclo(9-14)CRPRLC is a competitive antagonist at APJ. Radioligand binding in CHO cells transfected with human APJ fave a K(i) value of 82nM, competition binding in human left ventricle gave a K(D) value of 3.2 μM, and cAMP accumulation assays in CHO-K1-APJ cells fave a K(D) value of 1.32μM.
Macaluso NJ, Pitkin SL, Maguire JJ, Davenport AP, Glen RC. ChemMedChem. 2011 Jun 6;6(6):1017-23. doi: 10.1002/cmdc.201100069. Epub 2011 May 10.