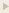|
|
|
AcSDKP |
Functional peptide, N-acetyl-seryl-aspartyl-lysyl-proline derived from Thymosin β4 |
Synthesis and metabolism of AcSDKP. Tβ4, a G-actying binding peptide, is cleaved by POP, and subsequently its N-terminal tetrapeptide AcSDKP, is synthesized. AcSDKP is hydrolysed and degraded by ACE. ACE-1 maty suppress miR-324-3p, which may inhibit protein expression of POP. Therefore, the mechanisms underlying the increased levels of AcSDKP by ACE-1 may include both the suppression of degradation pathway and the induction of synthesis of AcSDKP.
Figure from: Kanasaki K. et al., Front Pharmacol. 2014 Apr 14;5:70
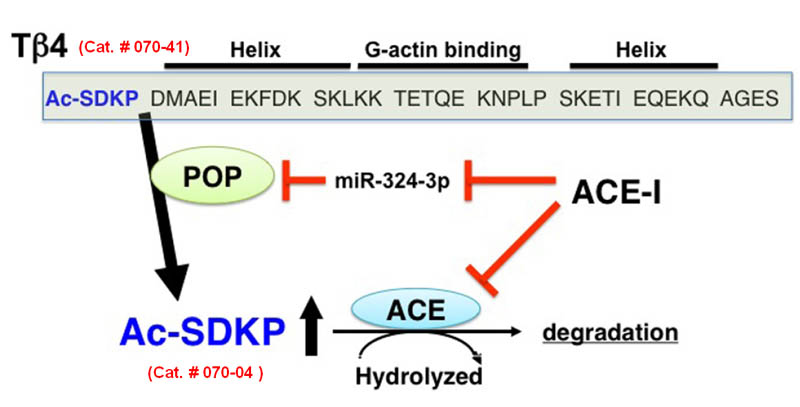
| Beneficial effects of AcSDKP in the process of tissue repair. AcSDKP exhibits multiple functions shown above, such as regulation of inflammation as well as anti-fibrotic, anti-apoptotic, and pro-angiogenic activities. Therefore, AcSDKP could be a candidate target molecule to combat kidney fibrosis in diabetes
Figure from: Kanasaki K. et al., Front Pharmacol. 2014 Apr 14;5:70 |
Fibroproliferative diseases are responsible for 45% of deaths in the developed world. Curing organ fibrosis is essential for fibroproliferative diseases. Diabetic nephropathy is a common fibroproliferative disease of the kidney and is associated with multiorgan dysfunction. However, therapy to combat diabetic nephropathy has not yet been established. In this review, we discuss the novel therapeutic possibilities for kidney fibrosis in diabetes focusing on the endogenous anti-fibrotic peptide, N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP), which is the substrate for angiotensin-converting enzyme and exhibits meaningful anti-fibrotic effects in various experimental models of fibrotic disease.
Kanasaki K, Nagai T, Nitta K et al., Front Pharmacol. 2014 Apr 14;5:70, 1-5. eCollection 2014.
Endothelial-to-mesenchymal transition (EndMT) emerges as an important source of fibroblasts. MicroRNA let-7 exhibits anti-EndMT effects and fibroblast growth factor (FGF) receptor has been shown to be an important in microRNA let-7 expression. The endogenous antifibrotic peptide N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) is a substrate of angiotensin-converting enzyme (ACE). Here, we found that AcSDKP inhibited the EndMT and exhibited fibrotic effects that were associated with FGF receptor-mediated anti-fibrotic program. Conventional ACE inhibitor plus AcSDKP ameliorated kidney fibrosis and inhibited EndMT compared to therapy with the ACE inhibitor alone in diabetic CD-1 mice. The endogenous AcSDKP levels were suppressed in diabetic animals. Cytokines induced cultured endothelial cells into EndMT; coincubation with AcSDKP inhibited EndMT. Expression of microRNA let-7 family was suppressed in the diabetic kidney; antifibrotic and anti-EndMT effects of AcSDKP were associated with the restoration of microRNA let-7 levels. AcSDKP restored diabetes- or cytokines-suppressed FGF receptor expression/phosphorylation into normal levels both in vivo and in vitro. These results suggest that AcSDKP is an endogenous antifibrotic molecule that has the potential to cure diabetic kidney fibrosis via an inhibition of the EndMT associated with the restoration of FGF receptor and microRNA let-7.
Nagai T, Kanasaki M, Srivastava SP et al., Biomed Res Int. 2014;2014:696475. doi: 10.1155/2014/696475. Epub 2014 Mar 24.
The primary limitation of thrombolytic treatment of ischemic stroke with tissue plasminogen activator (tPA) is the hemorrhagic risk. We tested AcSDKP (N-acetyl-seryl-aspartyl-lysyl-proline), as an auxiliary therapeutic agent, to reduce blood-brain barrier (BBB) disruption in a combination tPA thrombolytic treatment of stroke. Wistar rats subjected to embolic stroke were randomly assigned to either the tPA monotherapy group (n=9) or combination of tPA and AcSDKP treatment group (n=9) initiated at 4h after ischemia. Magnetic resonance imaging (MRI) measurements were performed before and after the treatments. Immunohistochemical staining and measurements were performed to confirm MRI findings. Longitudinal MRI permeability measurements with gadolinium-diethylenetriamine penta-acetic acid (Gd-DTPA) demonstrated that combination treatment of acute embolic stroke with AcSDKP and tPA significantly reduced BBB leakage, compared to tPA monotherapy, at 3 and 6days (18.3±9.8mm(3) vs 65.0±21.0mm(3), p<0.001) after the onset of stroke, although BBB leakage was comparable between the two groups prior to the treatments (6.8±4.4mm(3) vs 4.3±3.3mm(3), p>0.18). The substantial reduction of BBB leakage observed in the combination treatment group was closely associated with reduced ischemic lesions measured by T2 maps (113.6±24.9mm(3) vs 188.1±60.8mm(3), p<0.04 at 6days). Histopathological analysis of the same population of rats showed that the combination treatment significantly reduced parenchymal fibrin deposition (0.063±0.059mm(2) vs 0.172±0.103mm(2), p<0.03) and infarct volume (146.7±35.9mm(3) vs 199.3±60.4mm(3), p<0.05) compared to the tPA monotherapy at 6days after stroke. MRI provides biological insight into the therapeutic benefit of combination treatment of stroke with tPA and AcSDKP 4h after onset, and demonstrates significantly improved cerebrovascular integrity with neuroprotective effects compared with tPA monotherapy.
Ding G, Zhang Z, Chopp M, et al., Neuroscience. 2014 Jun 20;271:1-8. doi: 10.1016/j.neuroscience.2014.04.025. Epub 2014 Apr 24.
AIM:
To investigate the preventive effect of N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) on bile duct ligation (BDL)-induced liver fibrosis in rats.
METHODS:
Liver fibrosis in rats was induced by BDL and AcSDKP was infused subcutaneously for 2 wk via a osmotic minipump (Alzet 2ML4) immediately after BDL operation. After scarifying, serum and liver specimens were collected. Hematoxylin and eosin staining, Sirius red staining, enzyme linked immunosorbent assay, Western blot or real-time polymerase chain reaction were used to determinate liver functions, histological alterations, collagen deposition, mRNA expression of markers for fibroblasts, transforming growth factor-β1 (TGF-β1) and bone morphogenetic protein-7 (BMP-7).
RESULTS:
When compared to model rats, chronic exogenous AcSDKP infusion suppressed profibrogenic TGF-β1 signaling, α-smooth muscle actin positivity (α-SMA), fibroblast specific protein-1 (FSP-1) staining and collagen gene expression. Col I, Col III, matrix metalloproteinase-2, tissue inhibitors of metalloproteinase-1 and tissue inhibitors of metalloproteinase-2 mRNA expressions were all significantly downregulated by AcSDKP infusion (2.02 ± 1.10 vs. 14.16 ± 6.50, 2.02 ± 0.45 vs. 10.00 ± 3.35, 2.91 ± 0.30 vs. 7.83 ± 1.10, 4.64 ± 1.25 vs. 18.52 ± 7.61, 0.46 ± 0.16 vs. 0.34 ± 0.12, respectively, P < 0.05). Chronic exogenous AcSDKP infusion attenuated BDL-induced liver injury, inflammation and fibrosis. BDL caused a remarkable increase in alanine transaminase, aspartate transaminase, total bilirubin, and prothrombin time, all of which were reduced by AcSDKP infusion. Mast cells, collagen accumulation, α-SMA, TGF-β1, FSP-1 and BMP-7 increased. The histological appearance of liver specimens was also improved.
CONCLUSION:
Infusion of exogenous AcSDKP attenuated BDL-induced fibrosis in the rat liver. Preservation of AcSDKP may be a useful therapeutic approach in the management of liver fibrosis.
Zhang L, Xu LM, Chen YW et al., World J Gastroenterol. 2012 Oct 7;18(37):5283-8. doi: 10.3748/wjg.v18.i37.5283.
BACKGROUND:
Hypertension-induced renal injury is characterized by inflammation, fibrosis and proteinuria. Previous studies have demonstrated that N-acetyl-Ser-Asp-Lys-Pro (Ac-SDKP) inhibits renal damage following diabetes mellitus and antiglomerular basement membrane nephritis. However, its effects on low-renin hypertensive nephropathy are not known. Thus, we hypothesized that Ac-SDKP has renal protective effects on deoxycorticosterone acetate (DOCA)-salt hypertensive mice, decreasing inflammatory cell infiltration, matrix deposition and albuminuria.
METHOD:
We uninephrectomized 16-week-old C57BL/6J mice and treated them with either placebo, DCOA (10 mg/10 g body weight subcutaneous) and 1% sodium chloride with 0.2% potassium chloride in drinking water (DOCA-salt) or DOCA-salt with Ac-SDKP (800 μg/kg per day) for 12 weeks. We measured blood pressure, urine albumin, glomerular matrix, renal collagen content, monocyte/macrophage infiltration and glomerular nephrin expression.
RESULTS:
Treatment with DOCA-salt significantly increased blood pressure (P < 0.01), which remained unaltered by Ac-SDKP. Ac-SDKP decreased DOCA-salt-induced renal collagen deposition, glomerular matrix expansion and monocyte/macrophage infiltration. Moreover, DOCA-salt-induced increase in albuminuria was normalized by Ac-SDKP (controls, 10.8 ± 1.7; DOCA-salt, 41 ± 5; DOCA-salt + Ac-SDKP, 13 ± 3 μg/10 g body weight per 24 h; P < 0.001, DOCA-salt vs. DOCA-salt + Ac-SDKP). Loss of nephrin reportedly causes excess urinary protein excretion; therefore, we determined whether Ac-SDKP inhibits proteinuria by restoring nephrin expression in the glomerulus of hypertensive mice. DOCA-salt significantly downregulated glomerular nephrin expression (controls, 37 ± 8; DOCA-salt, 10 ± 1.5% of glomerular area; P < 0.01), which was partially reversed by Ac-SDKP (23 ± 4.0% of glomerular area; P = 0.065, DOCA-salt vs. DOCA-salt + Ac-SDKP).
CONCLUSION:
We concluded that Ac-SDKP prevents hypertension-induced inflammatory cell infiltration, collagen deposition, nephrin downregulation and albuminuria, which could lead to renoprotection in hypertensive mice.
Rhaleb NE1, Pokharel S, Sharma U, Carretero OA., J Hypertens. 2011 Feb;29(2):330-8. doi: 10.1097/HJH.0b013e32834103ee.
Myocarditis is commonly associated with cardiotropic infections and has been linked to development of autoimmunity. N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a naturally occurring tetrapeptide that prevents inflammation and fibrosis in hypertension and other cardiovascular diseases; however, its effect on autoimmune-mediated cardiac diseases remains unknown. We studied the effects of Ac-SDKP in experimental autoimmune myocarditis (EAM), a model of T cell-mediated autoimmune disease. This study was conducted to test the hypothesis that Ac-SDKP prevents autoimmune myocardial injury by modulating the immune responses. Lewis rats were immunized with porcine cardiac myosin and treated with Ac-SDKP or vehicle. In EAM, Ac-SDKP prevented both systolic and diastolic cardiac dysfunction, remodeling as shown by hypertrophy and fibrosis, and cell-mediated immune responses without affecting myosin-specific autoantibodies or antigen-specific T cell responses. In addition, Ac-SDKP reduced cardiac infiltration by macrophages, dendritic cells, and T cells, pro-inflammatory cytokines [interleukin (IL)-1α, tumor necrosis factor-α, IL-2, IL-17] and chemokines (cytokine-induced neutrophil chemoattractant-1, interferon-γ-induced protein 10), cell adhesion molecules (intercellular adhesion molecule-1, L-selectin), and matrix metalloproteinases (MMP). Ac-SDKP prevents autoimmune cardiac dysfunction and remodeling without reducing the production of autoantibodies or T cell responses to cardiac myosin. The protective effects of Ac-SDKP in autoimmune myocardial injury are most likely mediated by inhibition of 1) innate and adaptive immune cell infiltration and 2) expression of proinflammatory mediators such as cytokines, chemokines, adhesion molecules, and MMPs.
Nakagawa P, Liu Y, Liao TD et al., Am J Physiol Heart Circ Physiol. 2012 Nov 1;303(9):H1114-27. doi: 10.1152/ajpheart.00300.2011. Epub 2012 Aug 24.
Thymosin beta 4 (Tβ4), a G-actin sequestering peptide, improves neurological outcome in rat models of neurological injury. Tissue inflammation results from neurological injury and regulation of the inflammatory response is vital for neurological recovery. The innate immune response system which includes the Toll-like receptor (TLR) proinflammatory signaling pathway regulates tissue injury. We hypothesized that Tβ4 regulates the TLR proinflammatory signaling pathway. Since oligodendrogenesis plays an important role in neurological recovery, we employed an in-vitro primary rat embryonic cell model of oligodendrocyte progenitor cells (OPCs) and a mouse N20.1 OPC cell line to measure the effects of Tβ4 on the TLR pathway. Cells were grown in the presence of Tβ4 ranging from 25 to 100 ng/ml of (RegeneRx Biopharmaceuticals Inc, Rockville, MD) for 4 days. Quantitative real-time (Qrt) PCR data demonstrated that Tβ4 treatment increased expression of microRNA-146a (mir-146a), a negative regulator the TLR signaling pathway, in these two cell models. Western blot analysis showed that Tβ4 treatment suppressed expression of IL-1 receptor associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6), two proinflammatory cytokines of the TLR signaling pathway. Transfection of mir-146a into both primary rat embryonic OPCs and mouse N20.1 OPCs treated with Tβ4 demonstrated an amplification of myelin basic protein (MBP) expression and differentiation of OPC into mature MBP expressing oligodendrocytes. Transfection of anti-mir-146a nucleotides reversed the inhibitory effect of Tβ4 on IRAK1 and TRAF6 and decreased expression of MBP. These data suggest Tβ4 suppresses the TLR proinflammatory pathway by upregulating miR146a.
Santra M, Zhang ZG, Yang J et al., J Biol Chem. 2014 May 14. pii: jbc.M113.529966. [Epub ahead of print]
|
|
|
%070-04%;%070-41%;%070-75%;%070-76%
|
|
|