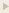N-formyl peptide receptors (FPRs) are critical regulators of host defense in phagocytes and are also expressed in epithelia. FPR signaling and function have been extensively studied in phagocytes, yet their functional biology in epithelia is poorly understood. We describe a novel intestinal epithelial FPR signaling pathway that is activated by an endogenous FPR ligand, annexin A1 (ANXA1), and its cleavage product Ac2-26, which mediate activation of ROS by an epithelial NADPH oxidase, NOX1. We show that epithelial cell migration was regulated by this signaling cascade through oxidative inactivation of the regulatory phosphatases PTEN and PTP-PEST, with consequent activation of focal adhesion kinase (FAK) and paxillin. In vivo studies using intestinal epithelial specific Nox1(-/-IEC) and AnxA1(-/-) mice demonstrated defects in intestinal mucosal wound repair, while systemic administration of ANXA1 promoted wound recovery in a NOX1-dependent fashion. Additionally, increased ANXA1 expression was observed in the intestinal epithelium and infiltrating leukocytes in the mucosa of ulcerative colitis patients compared with normal intestinal mucosa. Our findings delineate a novel epithelial FPR1/NOX1-dependent redox signaling pathway that promotes mucosal wound repair.
Leoni G, Alam A, Neumann PA et al., J Clin Invest. 2013 Jan 2;123(1):443-54. doi: 10.1172/JCI65831. Epub 2012 Dec 17.
In this study, we examined the therapeutic effects of an immune-stimulating peptide, WKYMVm, in ulcerative colitis. The administration of WKYMVm to dextran sodium sulfate (DSS)-treated mice reversed decreases in body weight, bleeding score and stool score in addition to reversing DSS-induced mucosa destruction and shortened colon. The WKYMVm-induced therapeutic effect against ulcerative colitis was strongly inhibited by a formyl peptide receptor (FPR) 2 antagonist, WRWWWW, indicating the crucial role of FPR2 in this effect. Mechanistically, WKYMVm effectively decreases intestinal permeability by stimulating colon epithelial cell proliferation. WKYMVm also strongly decreases interleukin-23 and transforming growth factor-β production in the colon of DSS-treated mice. We suggest that the potent immune-modulating peptide WKYMVm and its receptor FPR2 may be useful in the development of efficient therapeutic agents against chronic intestinal inflammatory diseases.
Kim SD, Kwon S, Lee SK et al., Exp Mol Med. 2013 Sep 13;45:e40. doi: 10.1038/emm.2013.77.
Endothelial colony-forming cells (ECFCs) are recruited to the sites of ischemic injury in order to contribute to neovascularization and repair of injured tissues. However, therapeutic potential of ECFCs is limited due to low homing and engraftment efficiency of transplanted ECFCs. The G-protein-coupled formyl peptide receptor (FPR) 2 has been implicated in regulation of inflammation and angiogenesis, while the role of FPR2 in homing and engraftment of ECFCs and neovascularization in ischemic tissues has not been fully defined. This study was undertaken to investigate the effects of WKYMVm, a selective FPR2 agonist isolated by screening synthetic peptide libraries, on homing ability of ECFCs and vascular regeneration of ischemic tissues. WKYMVm stimulated chemotactic migration, angiogenesis, and proliferation ability of human ECFCs in vitro. Small interfering RNA-mediated silencing of FPR2, but not FPR3, abrogated WKYMVm-induced migration and angiogenesis of ECFCs. Intramuscular injection of WKYMVm resulted in attenuation of severe hind limb ischemia and promoted neovascularization in ischemic limb. ECFCs transplanted via tail vein into nude mice were incorporated into capillary vessels in the ischemic hind limb, resulting in augmented neovascularization and improved ischemic limb salvage. Intramuscular injection of WKYMVm promoted homing of exogenously administered ECFCs to the ischemic limb and ECFC-mediated vascular regeneration. Silencing of FPR2 expression in ECFCs resulted in abrogation of WKYMVm-induced in vivo homing of exogenously transplanted ECFCs to the ischemic limb, neovascularization, and ischemic limb salvage. These results suggest that WKYMVm promotes repair of ischemic tissues by stimulating homing of ECFCs and neovascularization via a FPR2-dependent mechanism
Heo SC, Kwon YW, Jang IH et al., Stem Cells. 2014 Mar;32(3):779-90. doi: 10.1002/stem.1578.
Deficient wound healing in diabetic patients is very frequent, but the cellular and molecular causes are poorly defined. In this study, we have evaluated whether Annexin A1 derived peptide Ac2-26 stimulates fibroblast migration in high glucose conditions. Using normal human skin fibroblasts WS1 in low glucose (LG) or high glucose (HG) we observed the enrichment of Annexin A1 protein at cell movement structures like lamellipodial extrusions and interestingly, a significant decrease in levels of the protein in HG conditions. The analysis of the translocation of Annexin A1 to cell membrane showed lower levels of Annexin A1 in both membrane pool and supernatants of WS1 cells treated with HG. Wound-healing assays using cell line transfected with Annexin A1 siRNAs indicated a slowing down in migration speed of cells suggesting that Annexin A1 has a role in the migration of WS1 cells. In order to analyze the role of extracellular Annexin A1 in cell migration, we have performed wound-healing assays using Ac2-26 showing that peptide was able to increase fibroblast cell migration in HG conditions. Experiments on the mobilization of intracellular calcium and analysis of p-ERK expression confirmed the activity of the FPR1 following stimulation with the peptide Ac2-26. A wound-healing assay on WS1 cells in the presence of the FPR agonist fMLP, of the FPR antagonist CsH and in the presence of Ac2-26 indicated that Annexin A1 influences fibroblast cell migration under HG conditions acting through FPR receptors whose expression was slightly increased in HG. In conclusion, these data demonstrate that (i) Annexin A1 is involved in migration of WS1 cells, through interaction with FPRs; (ii) N- terminal peptide of Annexin A1 Ac2-26 is able to stimulate direct migration of WS1 cells in high glucose treatment possibly due to the increased receptor expression observed in hyperglycemia conditions.
Bizzarro V, Fontanella B, Carrat¨´ A et al, PLoS One. 2012;7(9):e45639. doi: 10.1371/journal.pone.0045639. Epub 2012 Sep 21.
BACKGROUND:
Recent studies suggest that the chemotactic G-protein-coupled-receptor (GPCR) formyl-peptide-receptor-like-1 (FPRL1) and the receptor-for-advanced-glycation-end-products (RAGE) play an important role in the inflammatory response involved in neurodegenerative disorders such as Alzheimer's disease (AD).Therefore, the expression and co-localisation of mouse formyl peptide receptor (mFPR) 1 and 2 as well as RAGE in an APP/PS1 transgenic mouse model using immunofluorescence and real-time RT-PCR were analysed. The involvement of rat or human FPR1/FPRL1 (corresponds to mFPR1/2) and RAGE in amyloid-β 1-42 (Aβ1-42)-induced signalling were investigated by extracellular signal regulated kinase 1/2 (ERK1/2) phosphorylation. Furthermore, the cAMP level in primary rat glial cells (microglia and astrocytes) and transfected HEK 293 cells was measured. Formyl peptide receptors and RAGE were inhibited by a small synthetic antagonist WRW4 and an inactive receptor variant delta-RAGE, lacking the intracytoplasmatic domains.
RESULTS:
We demonstrated a strong increase of mFPR1/2 and RAGE expression in the cortex and hippocampus of APP/PS1 transgenic mice co-localised to the glial cells. In addition, the Aβ1-42-induced signal transduction is dependant on FPRL1, but also on FPR1. For the first time, we have shown a functional interaction between FPRL1/FPR1 and RAGE in RAGE ligands S100B- or AGE-mediated signalling by ERK1/2 phosphorylation and cAMP level measurement. In addition a possible physical interaction between FPRL1 as well as FPR1 and RAGE was shown with co-immunoprecipitation and fluorescence microscopy.
CONCLUSIONS:
The results suggest that both formyl peptide receptors play an essential role in Aβ1-42-induced signal transduction in glial cells. The interaction with RAGE could explain the broad ligand spectrum of formyl peptide receptors and their important role for inflammation and the host defence against infections.
Slowik A, Merres J, Elfgen A et al., Mol Neurodegener. 2012 Nov 20;7:55. doi: 10.1186/1750-1326-7-55.
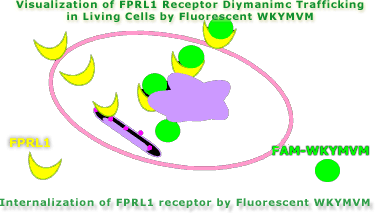
Neutrophils express the G protein-coupled N-formyl peptide receptor (FPR) and its homologue FPRL1, whereas monocytes express FPR, FPRL1, and FPRL2, an orphan receptor sharing 83% amino acid identity with FPRL1. FPRL1 is a promiscuous receptor activated by serum amyloid A and by different synthetic peptides, including the hexapeptide Trp-Lys-Tyr-Met-Val-d-Met-NH(2) (WKYMVm). By measuring calcium flux in HL-60 cells transfected with FPR, FPRL1, or FPRL2, we show that WKYMVm activated all three receptors, whereas the l-conformer WKYMVM activated exclusively FPRL1 and FPRL2. The functionality of FPRL2 was further assessed by the ability of HL-60-FPRL2 cells to migrate toward nanomolar concentrations of hexapeptides. The half-maximal effective concentrations of WKYMVM for calcium mobilization in HL-60-FPRL1 and HL-60-FPRL2 cells were 2 and 80 nm, respectively. Those of WKYMVm were 75 pm and 3 nm. The tritiated peptide WK[3,5-(3)H(2)]YMVM bound to FPRL1 (K(D) approximately 160 nm), but not to FPR. The two conformers similarly inhibited binding of (125)I-labeled WKYMVm to FPRL2-expressing cells (IC(50) approximately 2.5-3 micrometer). Metabolic labeling with orthophosphoric acid revealed that FPRL1 was differentially phosphorylated upon addition of the l- or d-conformer, indicating that it induced different conformational changes. In contrast to FPRL1, FPRL2 was already phosphorylated in the absence of agonist and not evenly distributed in the plasma membrane of unstimulated cells. However, both receptors were internalized upon addition of either of the two conformers. Taken together, the results indicate that neutrophils are activated by WKYMVM through FPRL1 and that FPRL2 is a chemotactic receptor transducing signals in myeloid cells.
Christophe T, Karlsson A, Dugave C et al., J Biol Chem. 2001 Jun 15;276(24):21585-93. Epub 2001 Apr 2.