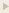|
|
|
FGF-23 receptor not only in kidney but also in vascular tissues |
Applications for C-terminal FGF-23 peptides |
(1) as a phosphaturic factor to induce hypophosphatemia, and low 1a,25(OH)2D3 synthesis with a reduction in the expression of 1a hydroxylase, leading to a negative P balance. Binding to Klotho protein to control mineral metabolism, vitamin D homeostasis, insulin/glucose, oxidative stress, and vascular calcifications.
Isakova T. et al, JAMA. 2011 Jun 15;305(23):2432-9.
(2) Fibroblast growth factor 23 acts on the parathyroid to decrease parathyroid hormone secretion.
Galitzer H, et al., Curr Opin Nephrol Hypertens. 2008 Jul;17(4):363-7.
(3) Increased level of FGF-23 has been linked to CVD and death of patients with Chronic Kidney Disease.
Wolf M. Clin J Am Soc Nephrol 5: 1727?729, 2010.

Action mechanisms of hormone-like Fgfs and regulatory mechanisms of their gene expression. (FGF-15) Intestinal FGF 15 expression is regulated by bile acid produced in the liver. The ligand-bound FXR forms a heterodimer with RXRs and induces the expression of FGF-15. The FGF-15 suppresses the expression of Cyp7a1 in the liver by activating the ßKlotho-Fgfr4 complex. The regulatory process forms a negative feedback loop in the regulation of bile acid homeostasis by FGF-15. (FGF-21) Hepatic FGF-21 expression is induced by the activation of PPARa. NEFA binds to and activates PPARa. The ligand-bound PPARa forms a heterodimer with RXRs and induces the expression of FGF-21. However, the regulatory mechanism of FGF-21 expression remains unclear. (FGF-23) Active vitamin D binds the vitamin D receptor (VDR). The ligand-bound VDR forms a heterodimer with retinoid X receptors (RXRs) and induces the expression of FGF-23. The increased FGF-23 suppresses the expression of Cyp27b1 and induces the expression of Cyp24 by activating the aKlotho-Fgfr1c complex. The regulatory process forms a negative feedback loop in the regulation of vitamin D homeostasis
Itoh N. et al, Cell Tissue Res. 2010, 342 (1):1-11.

FGF-23(180-251) (Catalog # 073-62) is a carboxyl-terminal peptide that is unique compared with other members of the FGF family.
BACKGROUND: Fibroblast growth factor (FGF)-23 levels have been associated with impaired vasoreactivity, increased arterial stiffness, and cardiovascular morbi-mortality, whereas a protective function of KLOTHO against endothelial dysfunction has been reported. Since expression of the FGF-23-KLOTHO system in human vascular tissue remains unproved, we aimed to study the expression of FGF-23, FGF receptors (FGFR) and KLOTHO in human aorta. In addition, we analyzed the FGF-23-KLOTHO expression in occlusive coronary thrombi.
METHODS: Thoracic aorta specimens from 44 patients underwent elective cardiac surgery, and thrombus material from 2 patients with acute coronary syndrome (ACS), were tested for FGF-23-KLOTHO system expression.
RESULTS: Expression of KLOTHO (mean expression level 4.85?.43, arbitrary units) and two of the three cognate FGFR (FGFR-1 and -3) were detected and confirmed by RT-PCR, sequencing and qRT-PCR. KLOTHO expression was confirmed within occlusive coronary thrombi from patients with ACS. However, expression of FGF-23 and FGFR4 was not observed. We also detected the aortic expression of membrane-anchored A Desintegrin and Metalloproteinases (ADAM)-17, the enzyme responsible for the shedding of KLOTHO from the cell surface, and the anti-inflammatory cytokine interleukin (IL)-10. Interestingly, in aortic samples there was a direct association between KLOTHO mRNA levels and those of ADAM-17 and IL-10 (r=0.54, P< 0.001; r=0.51, P< 0.01, respectively).
CONCLUSIONS: Human vascular tissue expresses members of the FGF-23-KLOTHO system, indicating that it can be a direct target organ for FGF-23. In addition, KLOTHO expression is also detected in occlusive coronary thrombi. These findings suggest a putative role of FGF-23-KLOTHO axis in human vascular pathophysiology and cardiovascular disease.
Donate-Correa J et al., Int J Cardiol. 2011 Sep 24
Fibroblast growth factor (FGF) 23 and Klotho are two factors associated with several metabolic disorders. Similar to humans, accelerated aging processes characterized by chronic vascular disease, bone demineralization, skin atrophy and emphysema have been recognized in FGF-23-null mice and Klotho-deficient mice. The role of these factors in the control of mineral metabolism homeostasis have been shown recently, particularly at the level of parathyroid cells and also in modulating active vitamin D production, two phenomena which are relevant in the presence of chronic kidney disease. In addition, the hormonal affect of circulating FGF-23 and Klotho proteins on vascular reactivity, either directly on endothelial cell functions or indirectly by modulating the brain endothelin-1-dependent sympathetic nervous system activity, has contributed to understanding their role in the pathophysiology of hypertension and atherosclerotic vasculopathies. Consequently, very recent clinical investigations seem to confirm the involvement of Klotho in modulating the severity and prognosis of human cardiovascular (CV) disorders and longevity. The present review reports data related to the possible interactive effects of Klotho and FGF-23 on the prognosis of renal and CV diseases.
Bernheim J & Benchetrit S, Nephrol Dial Transplant. 2011 Aug;26(8):2433-8. Epub 2011 May 4.
The functional interaction between fibroblast growth factor 23 (FGF-23) and Klotho in the control of vitamin D and phosphate homeostasis is manifested by the largely overlapping phenotypes of FGF-23- and Klotho-deficient mouse models. However, to date, targeted inactivation of FGF receptors (FGFRs) has not provided clear evidence for an analogous function of FGFRs in this process. Here, by means of pharmacologic inhibition of FGFRs, we demonstrate their involvement in renal FGF-23/Klotho signaling and elicit their role in the control of phosphate and vitamin D homeostasis. Specifically, FGFR loss of function counteracts renal FGF-23/Klotho signaling, leading to deregulation of Cyp27b1 and Cyp24a1 and the induction of hypervitaminosis D and hyperphosphatemia. In turn, this initiates a feedback response leading to high serum levels of FGF-23. Further, we show that FGFR inhibition blocks FGF-23 transcription in bone and that this is dominant over vitamin D-induced FGF-23 expression, ultimately impinging on systemic FGF-23 protein levels. Additionally, we identify FGF-23 as a specific target gene of FGF signaling in vitro. Thus, in line with FGF-23- and Klotho-deficient mouse models, our study illustrates the essential function of FGFRs in the regulation of vitamin D and phosphate levels. Further, we reveal FGFR signaling as a novel in vivo control mechanism for FGF-23 expression in bone, suggesting a dual function of FGFRs in the FGF-23/Klotho pathway leading to vitamin D and phosphate homeostasis.
Wöhrle S et al., J Bone Miner Res. 2011 Oct;26(10):2486-97. doi: 10.1002/jbmr.478
Background: FGF-23 and PTH inhibit phosphate transport in renal proximal tubules.
Results: FGF-23-mediated inhibition requires the PDZ-domain adaptor protein NHERF-1 and synergizes with PTH.
Conclusion: FGF-23 and PTH induce modifications of NHERF-1 by different but complimentary pathways.
Significance: The interaction between FGF-23 and PTH on renal phosphate transport may provide a therapeutic rational for treatment of patients with FGF-23-associated hyphosphatemia.
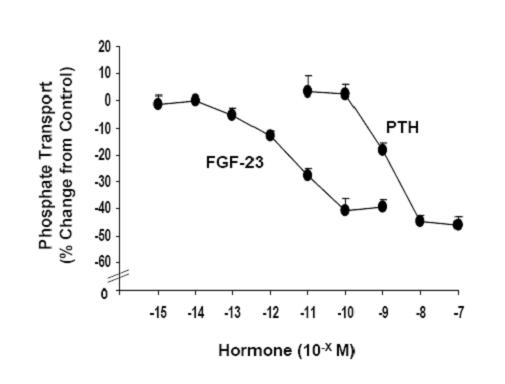
The dose-response relationship between FGF-23 and PTH, and phosphate transport was determined in BBM vesicles derived from wild-type mouse kidney slices incubated in the indicated concentration of hormone for 30 min. Non-hormone treated controls were obtained for each concentration tested and the results are expressed as Percent Change from Control ± SEM.
Three or more experiments were obtained with each concentration of hormone.
Weinman EJ. et al. Journal of Biological Chemistry Online Published on Sep 9, 2011
Fibroblast growth factor (FGF) 23 inhibits renal phosphate reabsorption by activating FGF receptor (FGFR) 1c in a Klotho-dependent fashion. The phosphaturic activity of FGF-23 is abrogated by proteolytic cleavage at the RXXR motif that lies at the boundary between the FGF core homology domain and the 72-residue-long C-terminal tail of FGF-23. Here, we show that the soluble ectodomains of FGFR1c and Klotho are sufficient to form a ternary complex with FGF-23 in vitro. The C-terminal tail of FGF-23 mediates binding of FGF-23 to a de novo site generated at the composite FGFR1c-Klotho interface. Consistent with this finding, the isolated 72-residue-long C-terminal tail of FGF-23 impairs FGF-23 signaling by competing with full-length ligand for binding to the binary FGFR-Klotho complex. Injection of the FGF-23 C-terminal tail peptide into healthy rats inhibits renal phosphate excretion and induces hyperphosphatemia. In a mouse model of renal phosphate wasting attributable to high FGF-23, the FGF-23 C-terminal peptide reduces phosphate excretion, leading to an increase in serum phosphate concentration. Our data indicate that proteolytic cleavage at the RXXR motif abrogates FGF-23 activity by a dual mechanism: by removing the binding site for the binary FGFR-Klotho complex that resides in the C-terminal region of FGF-23, and by generating an endogenous inhibitor of FGF-23. We propose that peptides derived from the C-terminal tail of FGF-23 or peptidomimetics and small-molecule organomimetics of the C-terminal tail can be used as therapeutics to treat renal phosphate wasting. (PS. FGF-23 (180-251) can compete with full length FGF-23 for binding to the FGFR-Klotho complex and antagonized the phosphaturic activity.)
Goetz R et al., Proc Natl Acad Sci U S A. 2010 Jan 5;107(1):407-12. Epub 2009 Dec 4.
|
|
|
FGF21;FGF-19;FGF2
%073-61%;%073-62%
|
|
|