|
An additional copy of the beta-amyloid precursor protein (APP) gene causes early-onset Alzheimer's disease (AD) in trisomy 21 (DS). Endosome dysfunction develops very early in DS and AD and has been implicated in the mechanism of neurodegeneration. Here, we show that morphological and functional endocytic abnormalities in fibroblasts from individuals with DS are reversed by lowering the expression of APP or beta-APP-cleaving enzyme 1 (BACE-1) using short hairpin RNA constructs. By contrast, endosomal pathology can be induced in normal disomic (2N) fibroblasts by overexpressing APP or the C-terminal APP fragment generated by BACE-1 (betaCTF), all of which elevate the levels of betaCTFs. Expression of a mutant form of APP that cannot undergo beta-cleavage had no effect on endosomes. Pharmacological inhibition of APP gamma-secretase, which markedly reduced Abeta production but raised betaCTF levels, also induced AD-like endosome dysfunction in 2N fibroblasts and worsened this pathology in DS fibroblasts. These findings strongly implicate APP and the betaCTF of APP, and exclude Abeta and the alphaCTF, as the cause of endocytic pathway dysfunction in DS and AD, underscoring the potential multifaceted value of BACE-1 inhibition in AD therapeutics.
Jiang, Y, et al. Proc Natl Acad Sci U S A. 2010 Jan 26;107(4):1630-5.
Intracellular amyloid-β peptide (Aβ) has been implicated in the pathogenesis of Alzheimer`s disease (AD). Mitochondria were found to be the target both for amyloid precursor protein (APP) that accumulates in the mitochondrial import channels and for Aβ that interacts with several proteins inside mitochondria and leads to mitochondrial dysfunction. Here, we have studied the role of mitochondrial γ-secretase in processing different substrates. We found that a significant proportion of APP is associated with mitochondria in cultured cells and that γ-secretase cleaves the shedded C-terminal part of APP identified as C83 associated with the outer membrane of mitochondria (OMM). Moreover, we have established the topology of the C83 in the OMM and found the APP intracellular domain (AICD) to be located inside mitochondria. Our data show for the first time that APP is a substrate for the mitochondrial γ-secretase and that AICD is produced inside mitochondria. Thus, we provide a mechanistic view of the mitochondria-associated APP metabolism where AICD, P3 peptide and potentially Aβ are produced locally and may contribute to mitochondrial dysfunction in AD.-Pavlov, P. F., Wiehager, B., Sakai, J., Frykman, S., Behbahani, H., Winblad, B., Ankarcrona, M. Mitochondrial γ-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein.
Pavlov PF, etc al. FASEB J. 2010 Sep 10
In recent decades, the study of the amyloid precursor protein (APP) and of its proteolytic products carboxy terminal fragment (CTF), APP intracellular C-terminal domain (AICD) and amyloid beta has been mostly focussed on the role of APP as a producer of the toxic amyloid beta peptide. Here, we reconsider the role of APP suggesting, in a provocative way, the protein as a central player in a putative signalling pathway. We highlight the presence in the cytosolic tail of APP of the YENPTY motif which is typical of tyrosine kinase receptors, the phosphorylation of the tyrosine, serine and threonine residues, the kinases involved and the interaction with intracellular adaptor proteins. In particular, we examine the interaction with Shc and Grb2 regulators, which through the activation of Ras proteins elicit downstream signalling events such as the MAPK pathway. The review also addresses the interaction of APP, CTFs and AICD with other adaptor proteins and in particular with Fe65 for nuclear transcriptional activity and the importance of phosphorylation for sorting the secretases involved in the amyloidogenic or non-amyloidogenic pathways. We provide a novel perspective on Alzheimer's disease pathogenesis, focussing on the perturbation of the physiological activities of APP-CTFs and AICD as an alternative perspective from that which normally focuses on the accumulation of neurotoxic proteolytic fragments.
Schettini G, et al. J Neurochem. 2010 Dec;115(6):1299-308.
The hypothesis that amyloid-β (Aβ) peptides are the primary cause of Alzheimer's disease (AD) remains the best supported theory of AD pathogenesis. Yet, many observations are inconsistent with the hypothesis. Aβ peptides are generated when amyloid precursor protein (APP) is cleaved by presenilins, a process that also produces APP intracellular domain (AICD). We previously generated AICD-overexpressing transgenic mice that showed abnormal activation of GSK-3β, a pathological feature of AD. We now report that these mice exhibit additional AD-like characteristics, including hyperphosphorylation and aggregation of tau, neurodegeneration and working memory deficits that are prevented by treatment with lithium, a GSK-3β inhibitor. Consistent with its potential role in AD pathogenesis, we find AICD levels to be elevated in brains from AD patients. The in vivo findings that AICD can contribute to AD pathology independently of Aβ have important therapeutic implications and may explain some observations that are discordant with the amyloid hypothesis.
Ghosal K., et al. PNAS, 2009,106 (43):18367-18372
The intracellular domain of the Alzheimer’s amyloid precursor protein (AICD) has been described as an important player in the transactivation of specific genes. It results from proteolytic processing of the Alzheimer’s amyloid precursor protein (APP), as does the neurotoxic Aβ peptide. Although normally produced in cells, Aβ is typically considered to be a neurotoxic peptide, causing devastating effects. By exposing primary neuronal cultures to relatively low Aβ concentrations, this peptide was shown to affect APP processing. Our findings indicate that APP C-terminal fragments are increased with concomitant reduction in the expression levels of APP itself. AICD nuclear immunoreactivity detected under control conditions was dramatically reduced in response to Aβ exposure. Additionally, intracellular protein levels of Fe65 and GSK3 were also decreased in response to Aβ. APP nuclear signaling is altered by Aβ, affecting not only AICD production but also its nuclear translocation and complex formation with Fe65. In effect, Aβ can trigger a physiological negative feedback mechanism that modulates its own production.
A. G. Henriques et al. J Mol Neurosci. 2009 September; 39(1-2): 248–255.
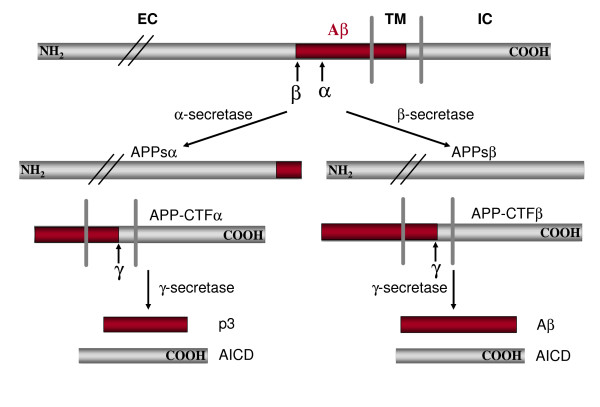
Schematic diagram of APP sequential processing (not drawn in scale). EC: extracellular; TM: transmembrane; IC: intracellular. Aβ domain is highlighted in red. For simplicity, only one cleavage site is shown for each enzyme. Hui Zheng and Edward H Koo, Mol Neurodegener. 2006; 1 : 5.
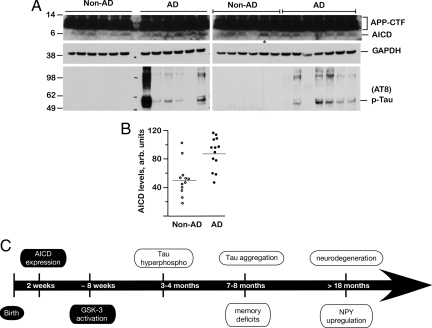
AICD levels are increased in human AD brains. (A) Brain lysates from 13 AD patients (AD) and 12 non-dementia control subjects (Non-AD) were analyzed by SDS/PAGE and Western blotted with 0443 antibody (top rows) that recognizes APP-C terminus or AT-8 antibody (bottom rows). Blots were stripped and reprobed with GAPDH (middle rows). The upper rows show APP-CTF and AICD levels, which were quantified by NIH ImageJ software (B). Although there was a large variation in the AICD levels, they were significantly elevated in AD patients compared to non-demented controls (P = 0.0017). The lane marked with * in A (right panel) marks the non-AD sample with abnormally high AICD levels (an outlier). The wide variation in AD samples is also seen in phospho-tau levels. (C) Summary of pathological events observed in FeCγ25 mice. AICD expression is driven by a CaMKIIα promoter, which becomes active around P15, and activation of GSK-3β is observed at 6–8 weeks. Increased phosphorylation of tau is first observed at 3–4 months of age, although tau does not become aggregated until 7–8 months of age. Memory deficits become first apparent at this time point. Neuronal loss and up-regulation of NPY are not observed up to 12–15 months of age but become apparent >18 months of age. AD-related pathological features described in this study are shown in open ovals.
Ghosal K., et al. PNAS, 2009,106 (43):18367-18372

|